|
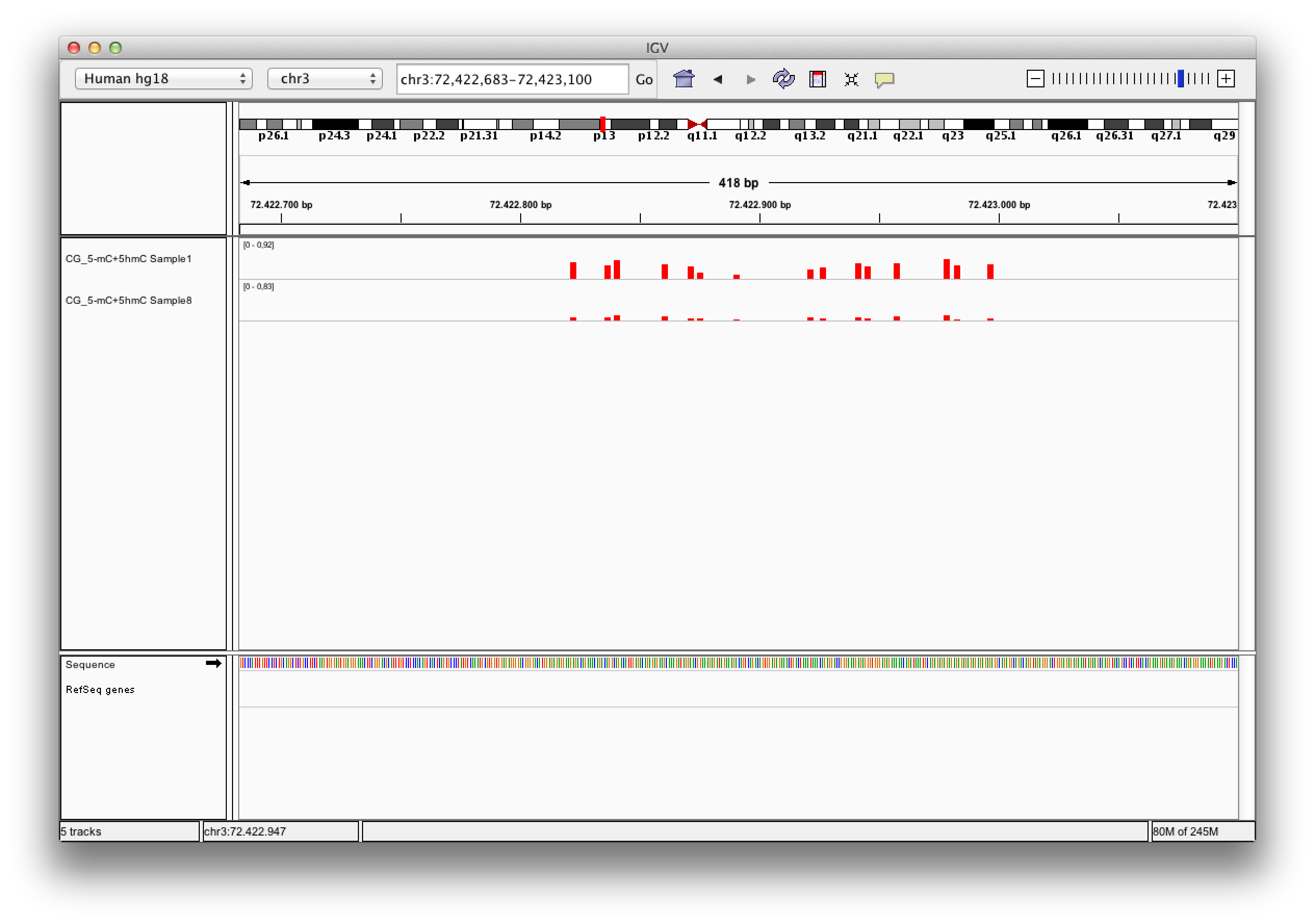
Data files returned from a sequencing machine cannot be used with BiQ Analyzer Himod
directly and have to be preprocessed. To prepare the data we
recommend a selection of tools included in Galaxy and provide a step by step guideline
on how to use them.
Galaxy
Galaxy can be used in various ways. We recommend using the main public Galaxy server which
can be accessed at https://usegalaxy.org/
Other options to use Galaxy are a local installation, a cloud instance,
using it on Slipstream or using one of the many other server based versions. All these
options can be choosen at http://galaxyproject.org/
In this tutorial we describe the usage of the main public server option.
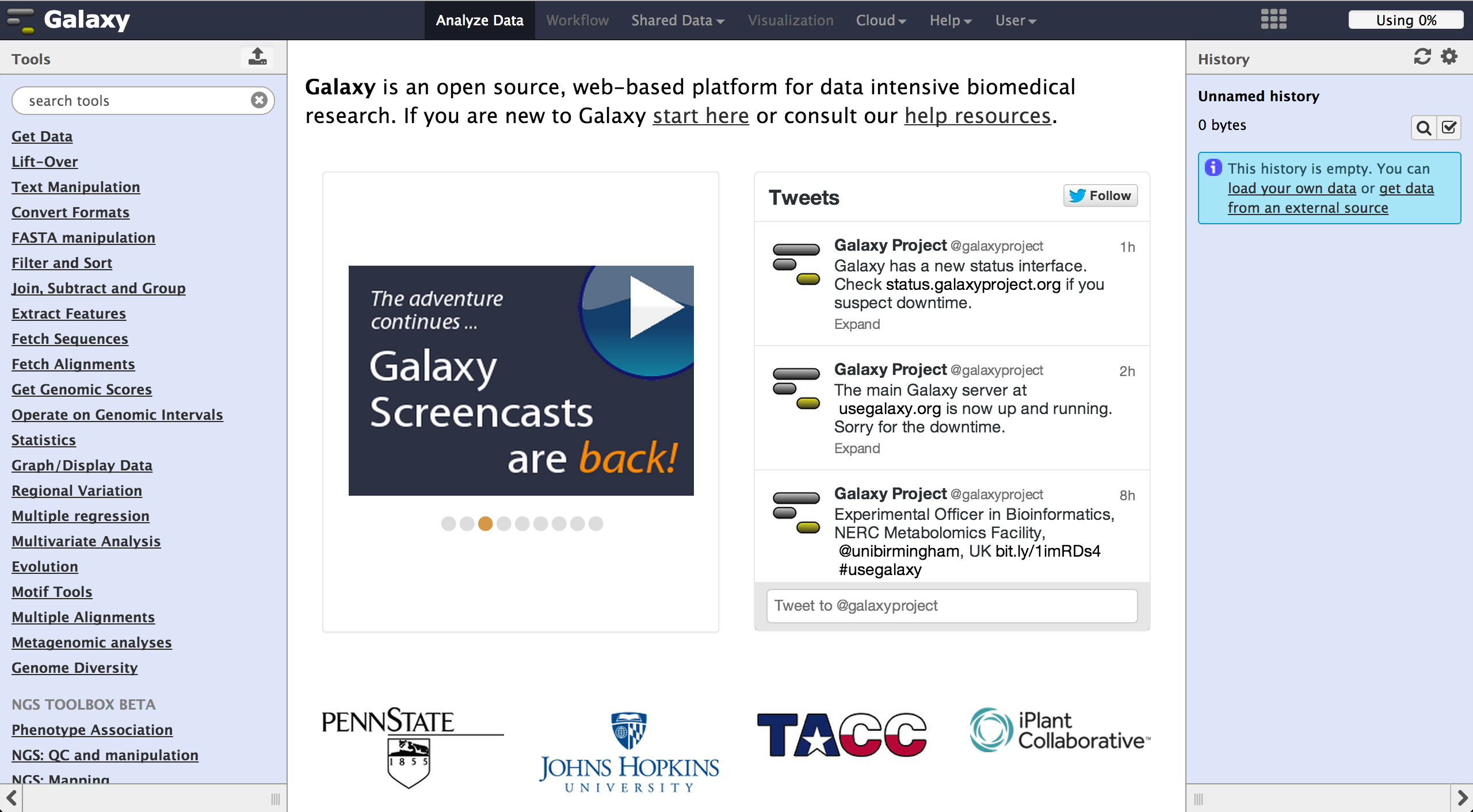
After opening Galaxy, one can see a list of tools on the left side and a history of
loaded and processed files on the right. The space in the middle shows the currently
open file or result.
|
|
 |
|
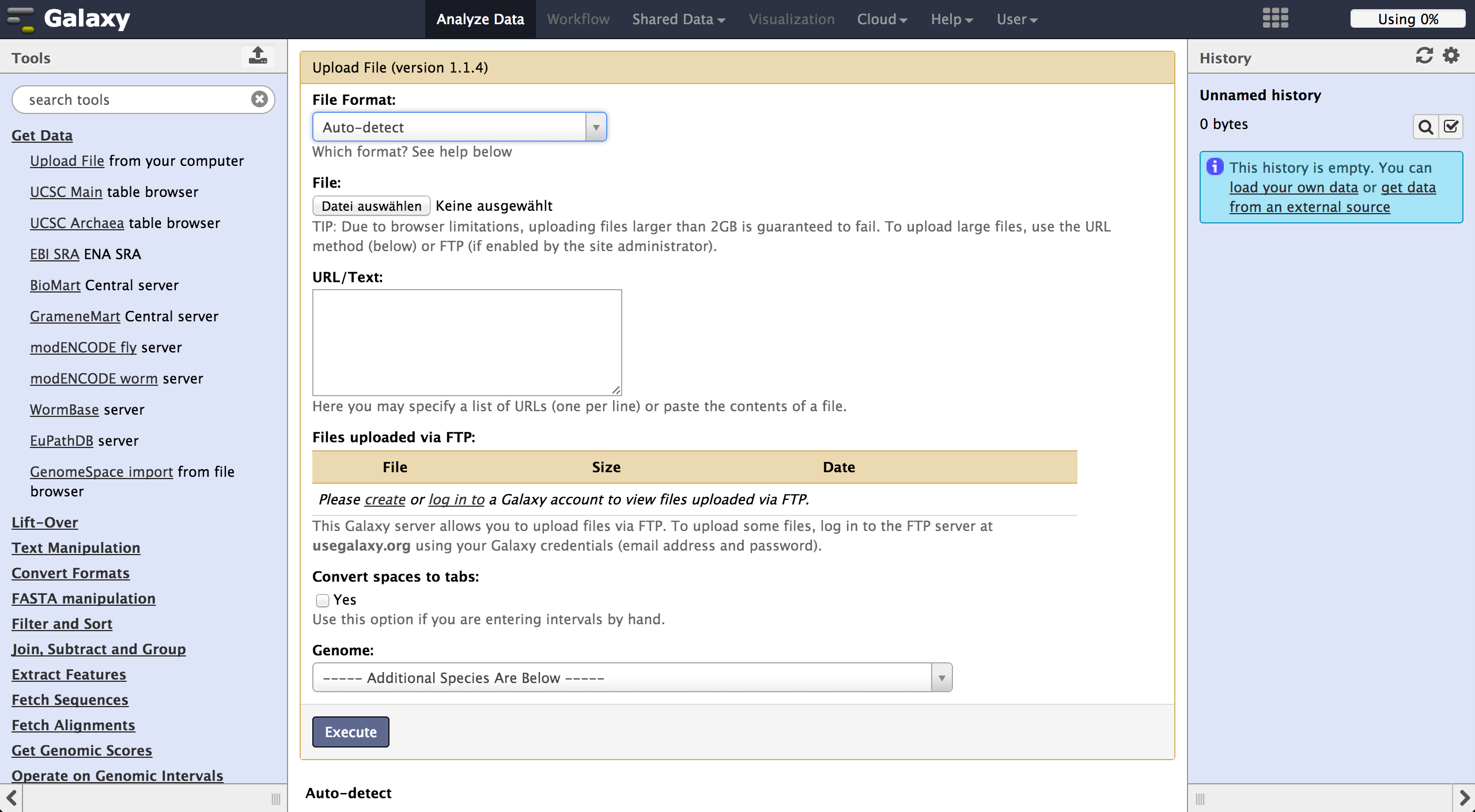
Loading files
To load files the user can choose "Get Data" -> "Upload File". The file format should be
automatically detected but the user can specify it if necessary. Files can be uploaded
from local harddisc, by URL or via a FTP Server. The user can further specify the genome
of their reads and let the tool automatically convert spaces to tabs while uploading the
file.
Uploaded files appear consecutively numbered in the history on the right side an can
be accessed there.
|
|
 |
|
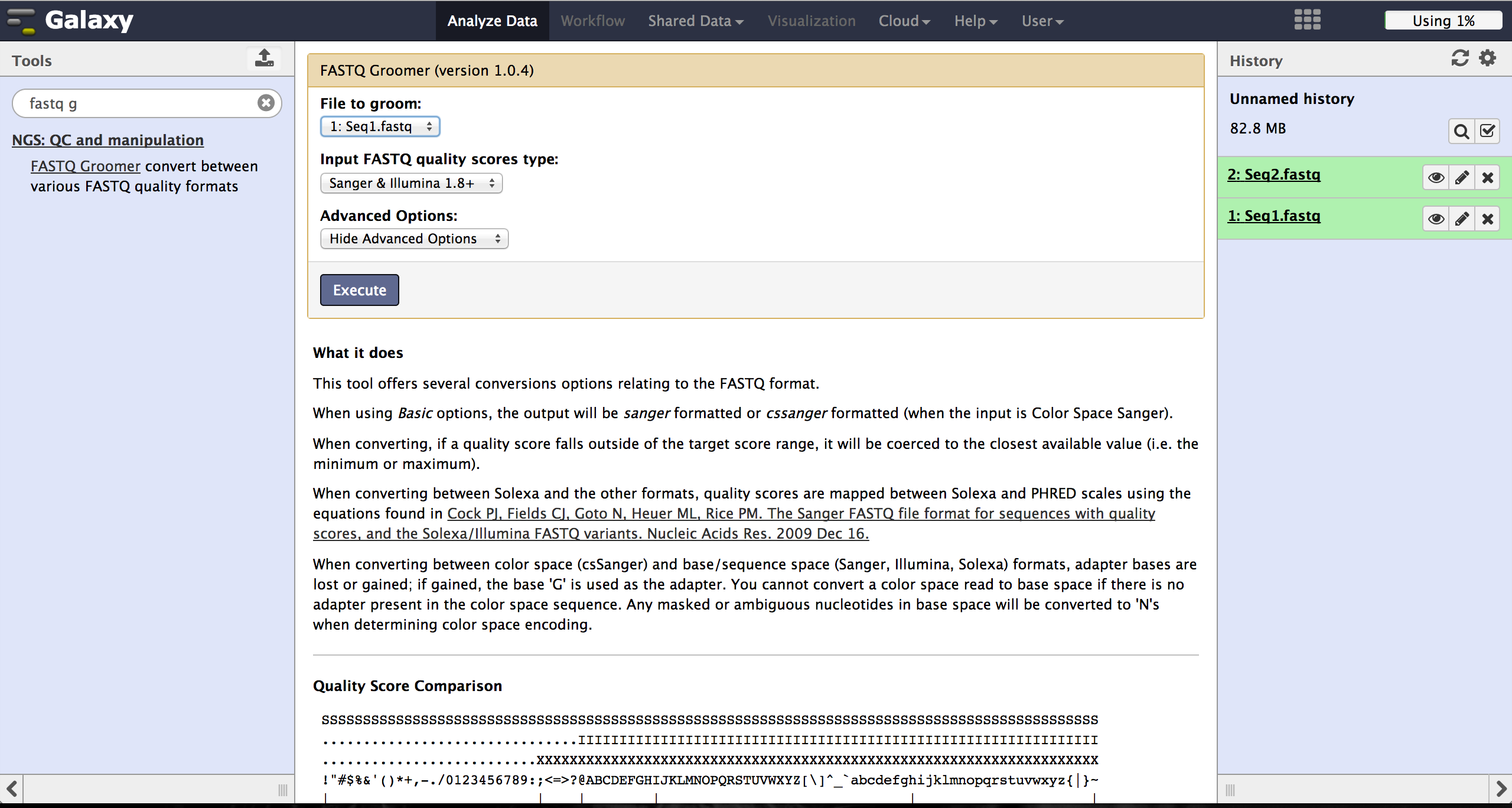
FastQ quality encoding - FASTQ Groomer
BiQ Analyzer HiMod's quality filter supports only Sanger or Illumina 1.8+ encoding. The
FASTQ Groomer can be used to convert encodings. This tool can be found at
"NGS: QC and manipulation" -> "FASTQ Groomer". Setting a loaded file and a given encoding
style, the FASTQ Groomer will change the encoding to Sanger, if nothing else is specified
in the advanved options. If the loaded file is already in Sanger format, it will not be
changed.
After using the FASTQ Groomer on a file, a new file with a name containing
"[]FASTQ Groomer on data[]" will appear in the history.
In case of working with paired end sequencing data, it is necessary to use this tool
as a first step. The clipping tool is very sensitive aboute quality encoding and will
only work on Sanger files.
|
|
 |
|
Clipping Adapter sequences - Clip
In case of a sequencing approach with paired ends, the user will end up with two files
per lane. One contains the reads started at the 5'end and the other the reads started
at the 3' end.
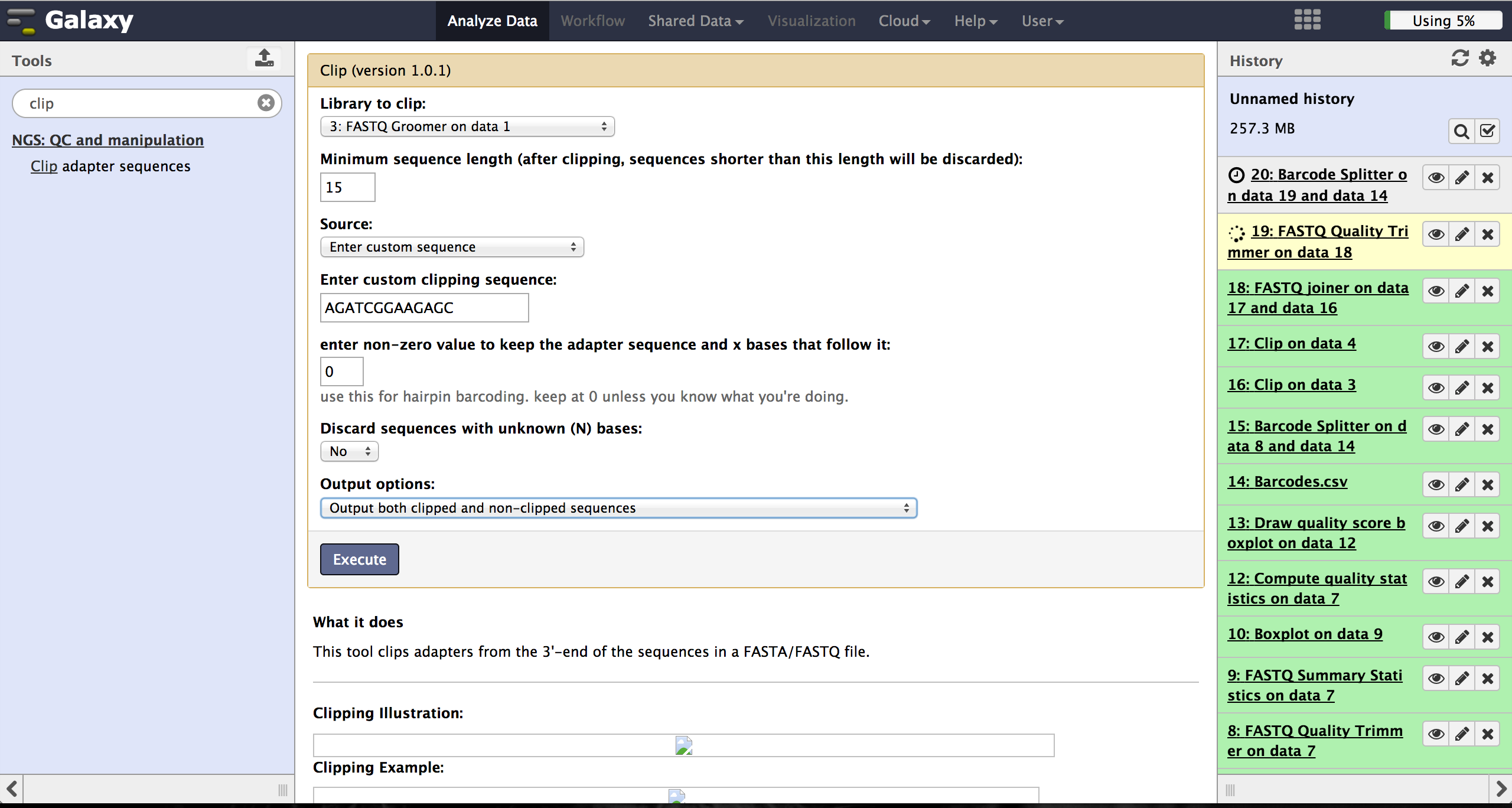
The Clip tool - accessible at "NGS: QC and manipulation" -> "Clip" - removes adapter
sequences to prepare the joining step. The user has to enter a groomed file and
choose an adapter sequence. The standard Illumina adapter sequence is
"AGATCGGAAGAGC". It is further necessary to specify which reads should be kept and which
should be discarded based on the clipping results.
|
|
 |
|
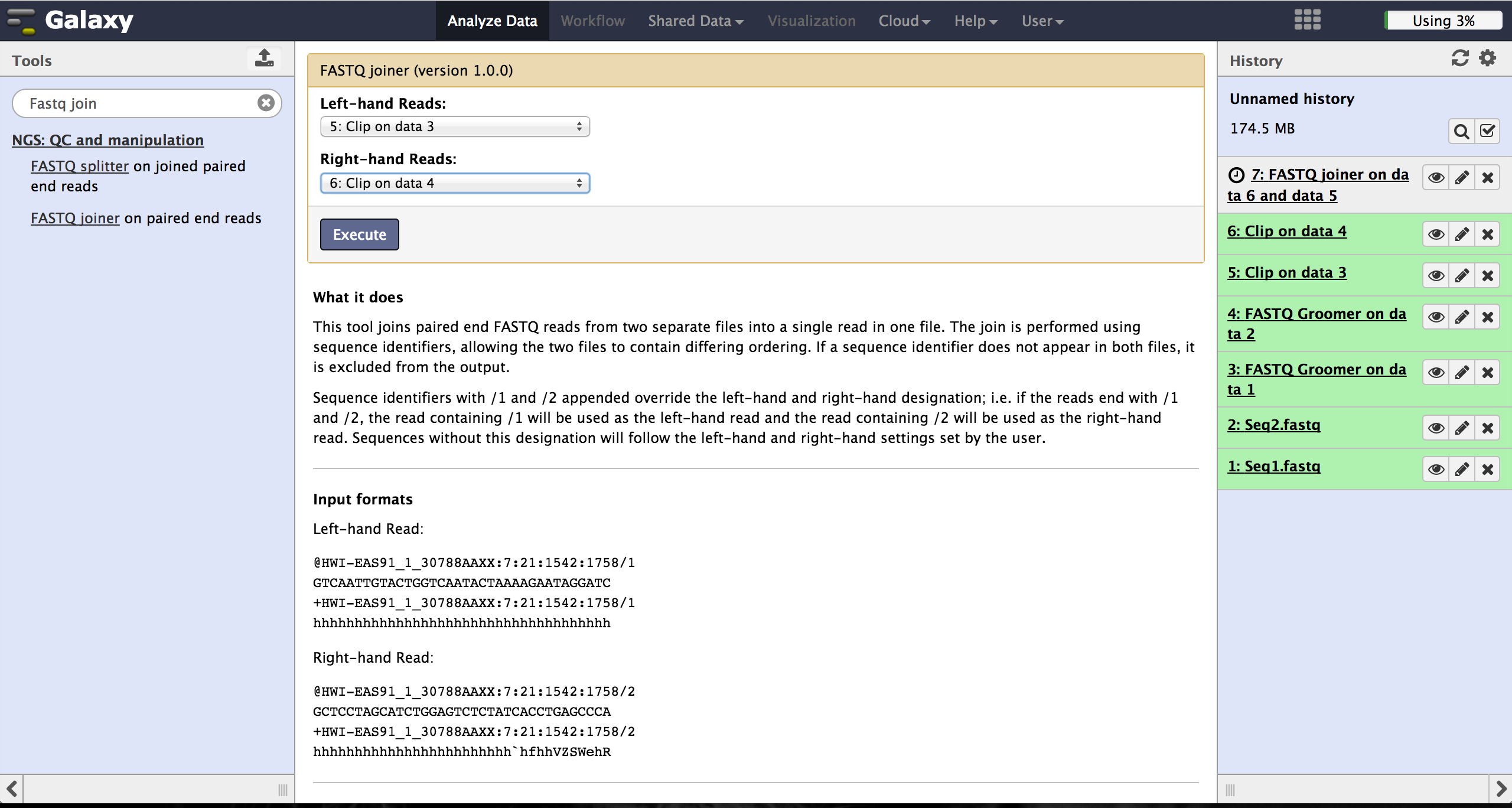
Joining paired reads - FASTQ joiner
The FASTQ joiner - accessible at "NGS: QC and manipulation" -> "FASTQ joiner" -
can join two files of a paired end sequencing and produce one file containing single
end reads. Therefore the user has to specify the two matching files for each lane and
run the tool.
|
|
 |
|
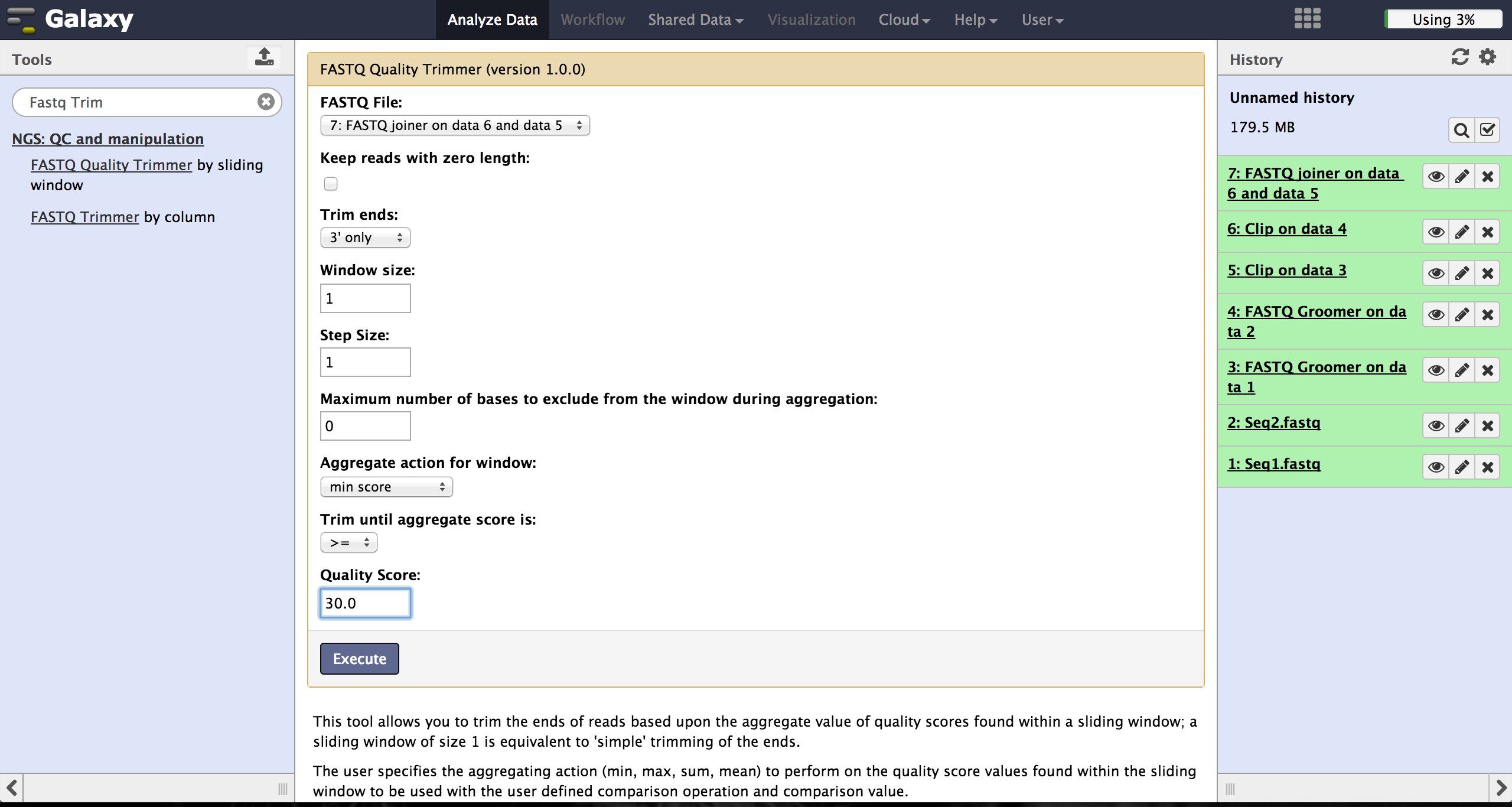
Trimming bad quality bases at read ends - FASTQ Quality Trimmer
The user can choose to trim reads that have ends with bad quality scores. The
FASTQ Quality Trimmer tool ("NGS: QC and manipulation" -> "FASTQ Quality Trimmer") uses
a sliding window approach to check for bases with bad quality and trims them until he
reaches an area with a good quality.
The user hast o specifiy the current FastQ file and a minimal quality score that
should be reached. Window size, step size and other parameters can also be set, but the
default parameters lead to usable results. One should only trim from the 3' end, because
the 5' end contains the primer sequences needed to demultiplex later on and should therefore not
be altered.
The 5' end can be trimmed in the end after demultiplexing.
|
|
 |
|
Trimming bad quality bases at read ends - FASTQ Trimmer
The FASTQ Trimmer ("NGS: QC and manipulation" -> "FASTQ Trimmer") deletes a specified
number of bases from each end of every read. This is another more strict method to get rid
of bad quality bases but comes with a loss of good bases at the same position. We
recommend the method using the FASTQ Quality Trimmer tool described above.
To find out a good parameter for each end, the user has to use FASTQ Summary
Statistics first. This can be found at "NGS: QC and manipulation" -> "FASTQ joiner".
Passing the joined FastQ file to this toll and executing it results in a table file
that can be used to produce a boxplot using Boxplot. Boxplot can be found at
"Graph/Display Data" -> "Boxplot". The user has to choose the Summary Statistics file and
start the tool. All other parameters can be left untouched.
By manually inspecting the boxplot, the user can decide which bases need to be
trimmed to reach a good enough quality score.
The results for 5'end and 3'end can be passed to the FASTQ Trimmer as well as the
current FastQ file. The run of the tool will result in a new trimmed FastQ file.
In this case the user should also only trim from the 3' end to not alter the primer sequence
on the 5' end.
|
|
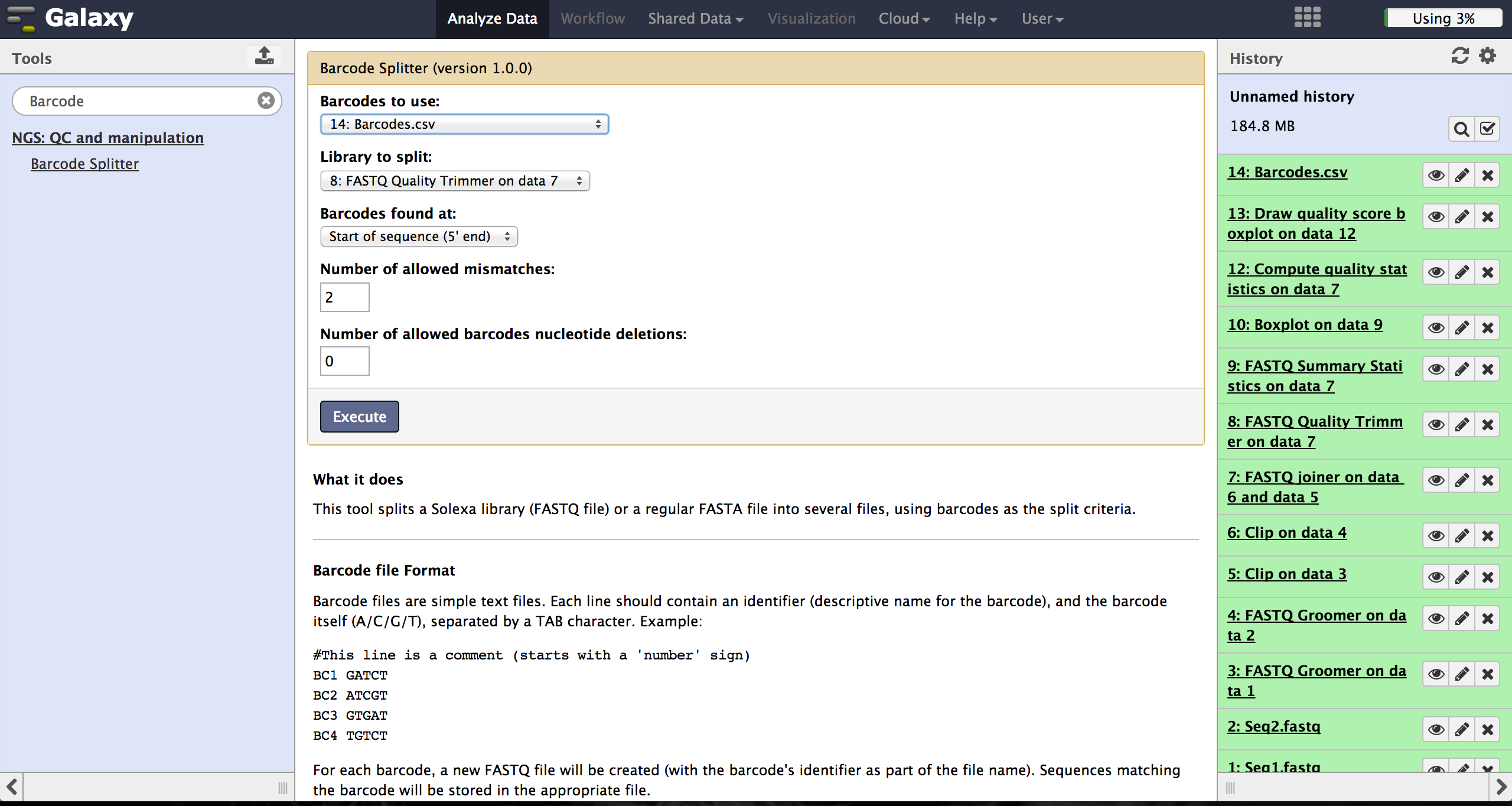
Demultiplexing - Barcode Splitter Version 1.0.0
As a last step it is necessary to demultiplex the FastQ file, if more than one amplicon
was sequenced per lane. To do so the user has to upload another file containing the
primers - which act as barcodes in this case - belonging to the different amplicons.
Such a file should be tab seperated and
contain in each line an identifier for the amplicon and the primer. All primers have to
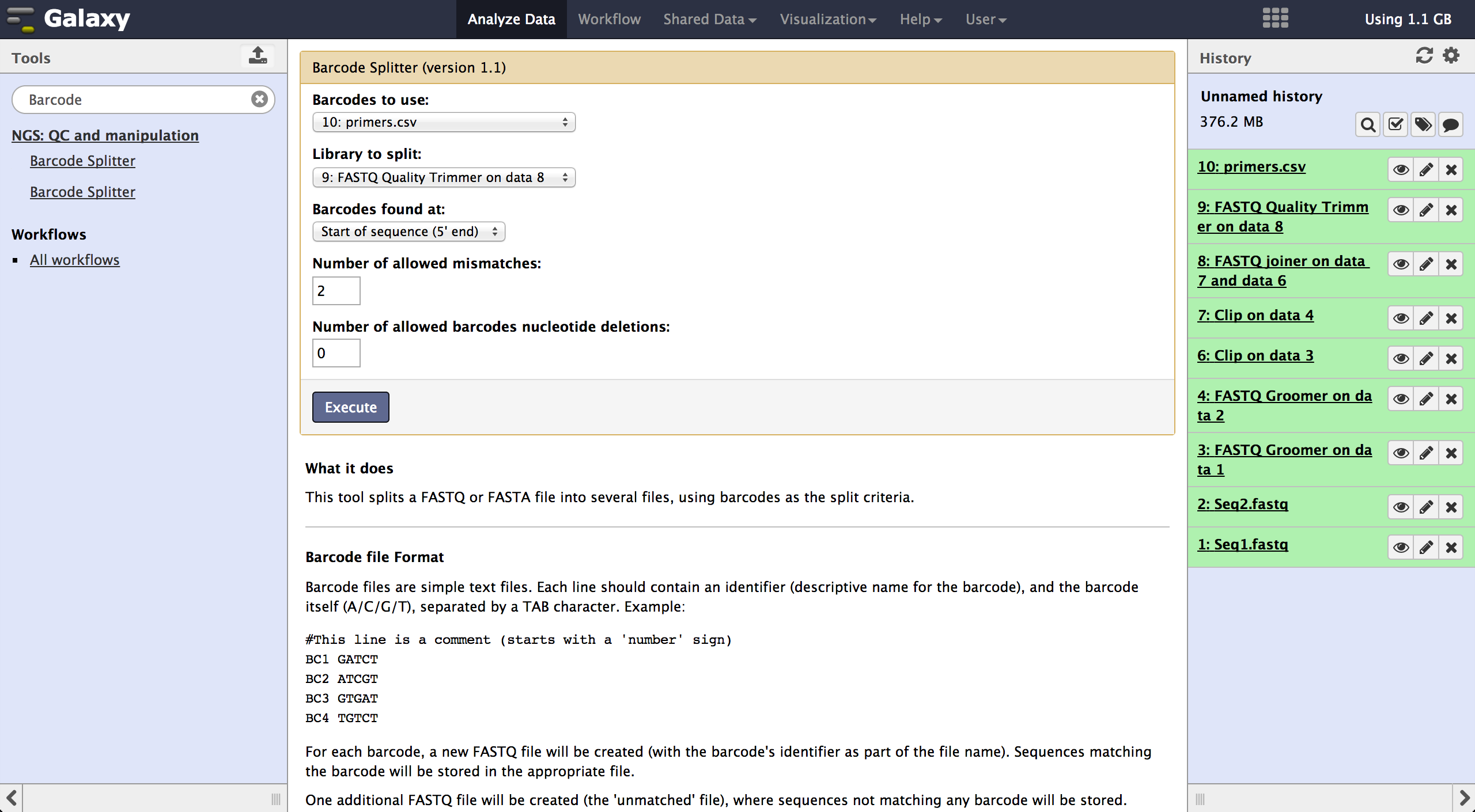
have the same length. The Barcode Splitter 1.0.0 can be found at "NGS: QC and manipulation"
-> "Barcode Splitter".
Specifying the barcode file, the current FastQ file and at which end of the reads the
primers can be found, the user can start the demultiplexing. The number of allowed
mismatches and the number of allowed primer nucleotide deletions can be given to
improve the results.
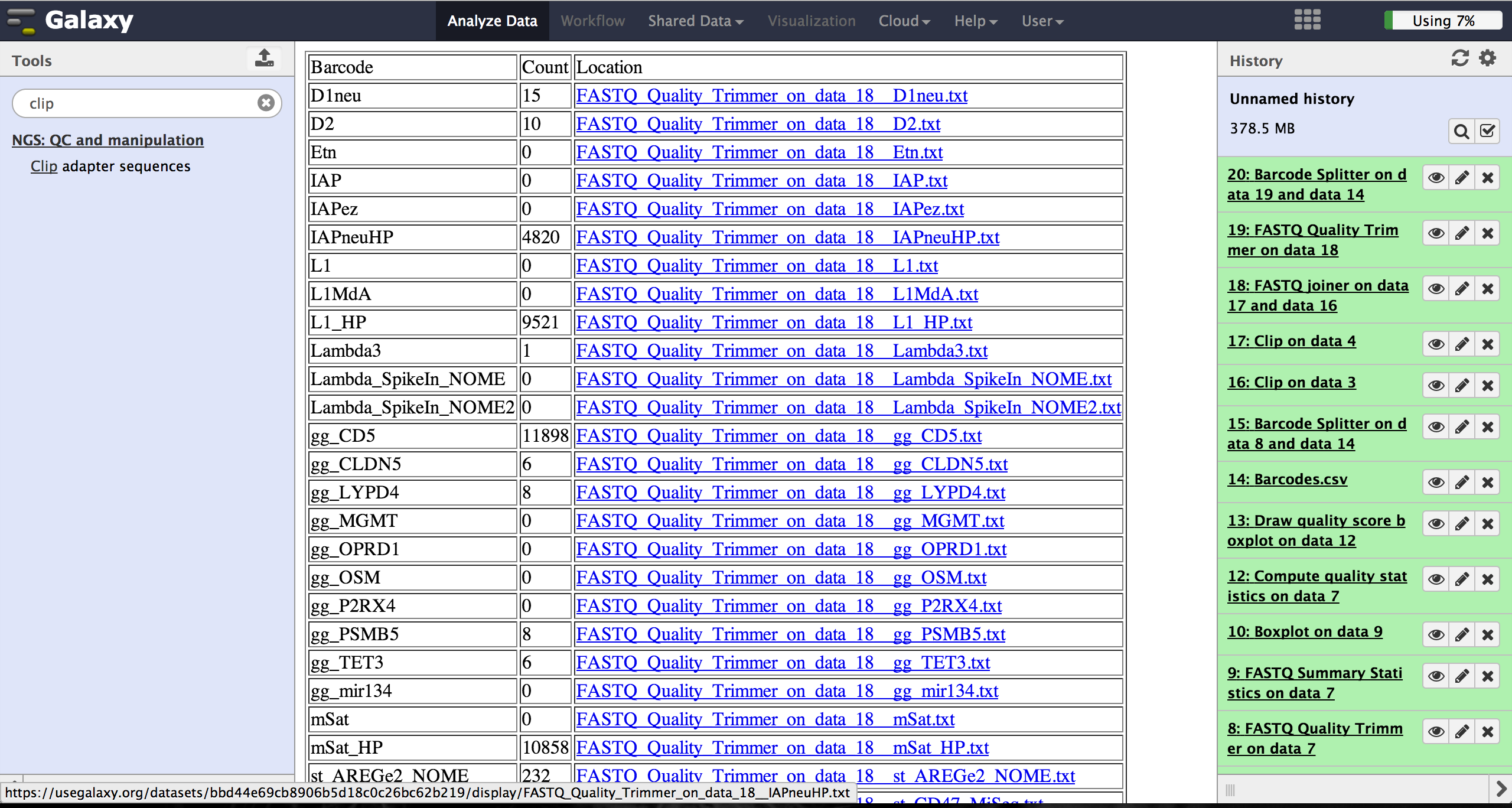
A new file appears in the history containing a table of the different identifiers,
how many reads mached this identifier and a link to a file containing these reads.
Opening such a file and clicking the download button on the demultiplexing result
file in the history downloads a zip archive including all new FastQ files. The filename
extension of those is txt but they contain fastq data and can therefore be easily renamed.
Sequencing platforms as Illumina MiSeq and HiSeq demultiplex by barcodes itself resulting
in fastq files, each containing only reads for one barcode per amplicon. The barcode is
given in the first line of each read. In case the raw
read files come from a platform which does not do this automatically, this step must be
repeated for all files resulting from the first demultiplexing step.
Otherwise if an own
version of Galaxy is used, it is possible to use another barcode splitter to perform
the demultiplexing.
|
|
 |
|
 |
|
Enhanced Demultiplexing - Barcode Splitter Version 1.1
This tool is not available on the main Galaxy server. To use it one has to run a separate
version of galaxy and install this tool to this version. In the galaxy toolshed it
is called "fastx_barcode_splitter_enhanced".
This tool might be very helpful if the user wants to modify the demultiplexed fastq files
further or demultiplex more than one time. It can be found at "NGS: QC and manipulation"
-> "FASTQ Trimmer". The only visible difference to the other barcode splitter is the
version number which is here 1.1.
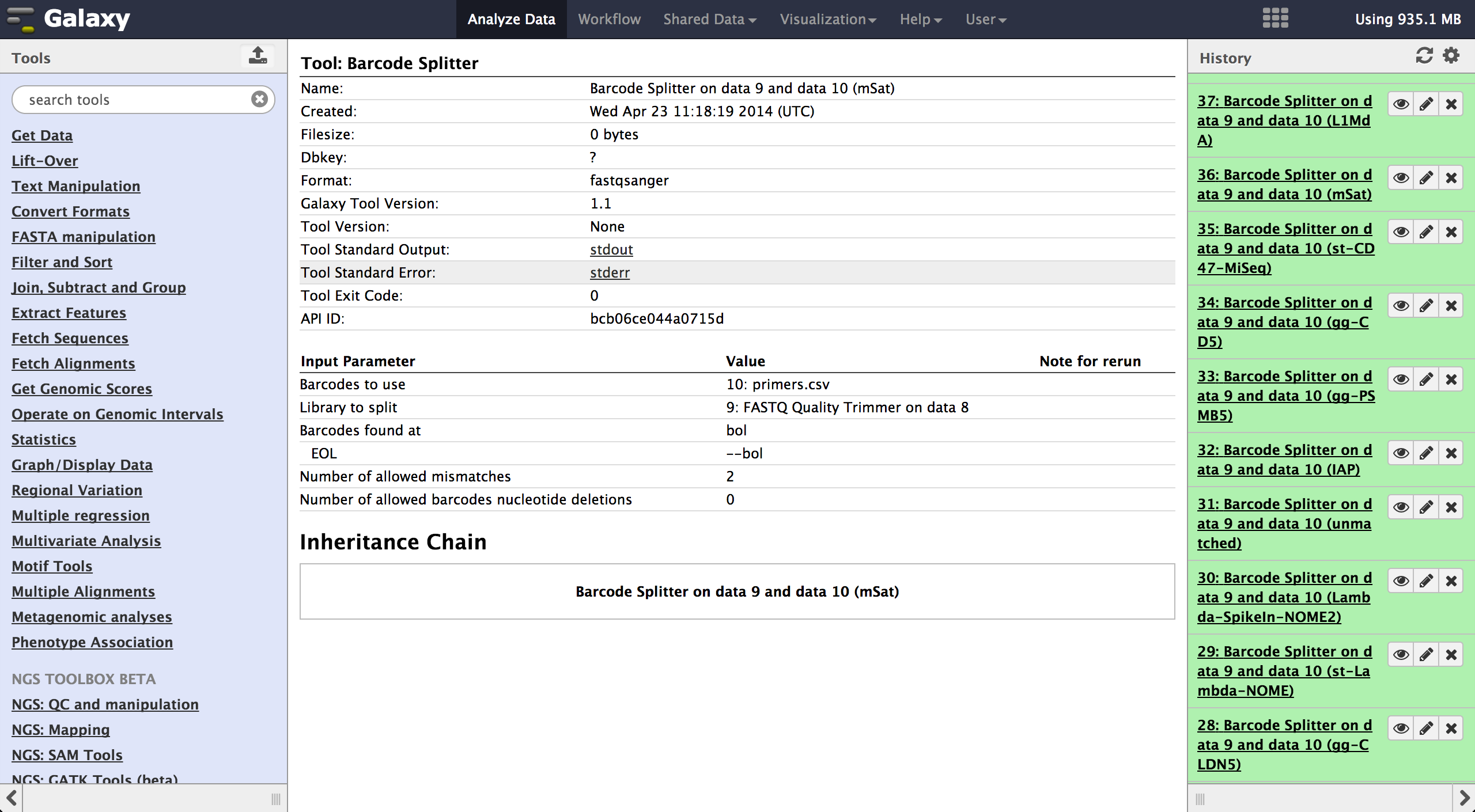
The tool is used exactly like the Barcode Splitter 1.0.0. The difference is, that every
result file is added to the galaxy history and can be further modified. Therefore it is
easy to consecutively perform multiple demultiplexing steps. The produced files can be
downloaded one by one.
|
|
 |
|
 |
|
Creating a spreadsheet
To easily load the created files into BiQ Analyzer HiMod, it is helpful to create a
spreadsheet containing the paths to all the files.
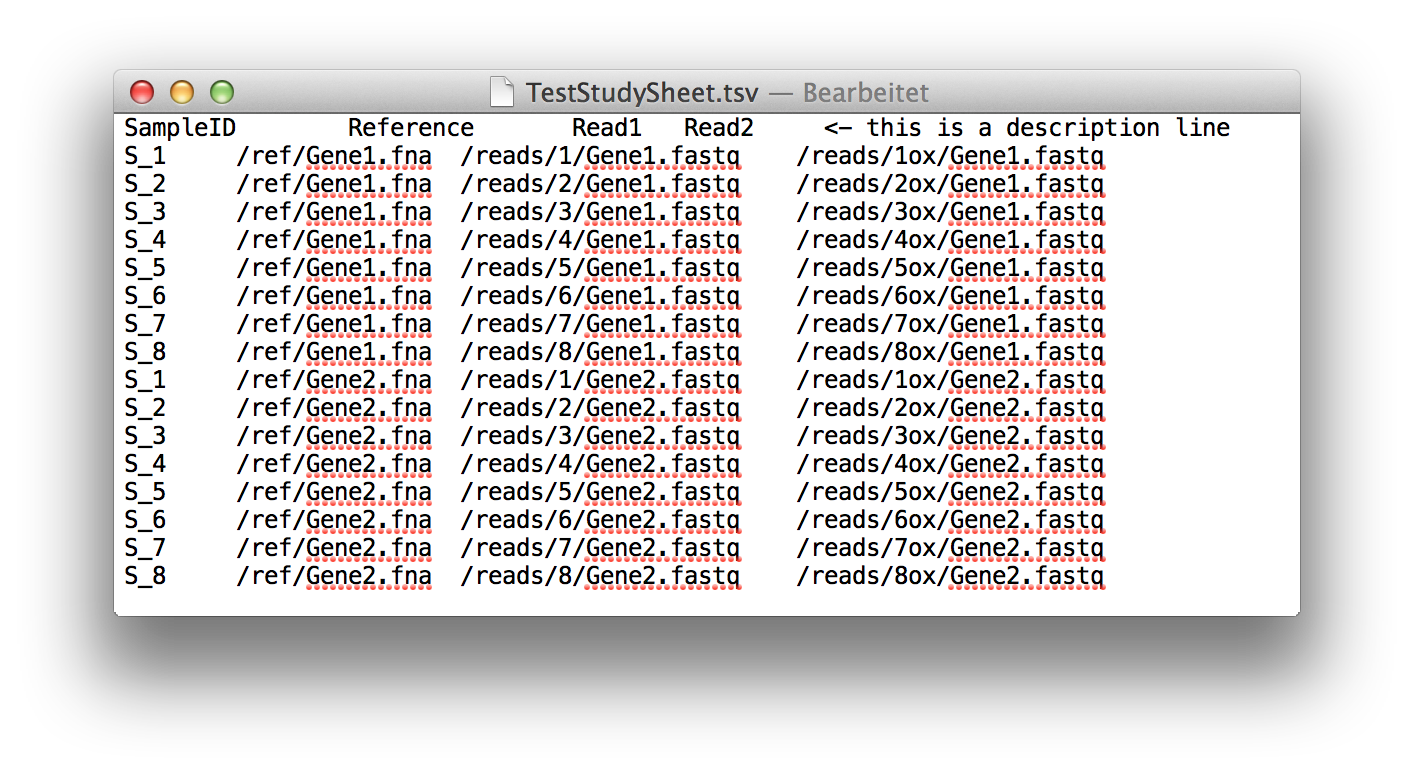
Such a spreadsheet starts with one line for comments and description. If this line is
empty, it will be ignored.
The following lines need to be tab-delimited and contain a sample name, the path to a
reference file and the paths to one or two read files, depending on the kind of analysis
that should be done.
Each possible combination of sample and amplicon needs to be listed in
one independent line. In case there are no reads for a probe, the according line can end
after the reference.
|
|
 |
|
References
- Goecks, J, Nekrutenko, A, Taylor, J and The Galaxy Team.
Galaxy: a comprehensive approach for supporting accessible, reproducible, and
transparent computational research in the life sciences. Genome Biol. 2010 Aug 25;11(8):R86.
- Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A,
Taylor J. "Galaxy: a web-based genome analysis tool for experimentalists". Current
Protocols in Molecular Biology. 2010 Jan; Chapter 19:Unit 19.10.1-21.
- Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D,
Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A. "Galaxy: a platform for interactive
large-scale genome analysis." Genome Research. 2005 Oct; 15(10):1451-5.
|